Rare genetic disorder linked to liver dysfunction, hypoglycemia, lipodystrophy
Mutation in ACAA2 gene modifies how body breaks down fat for fuel, UTSW researchers find
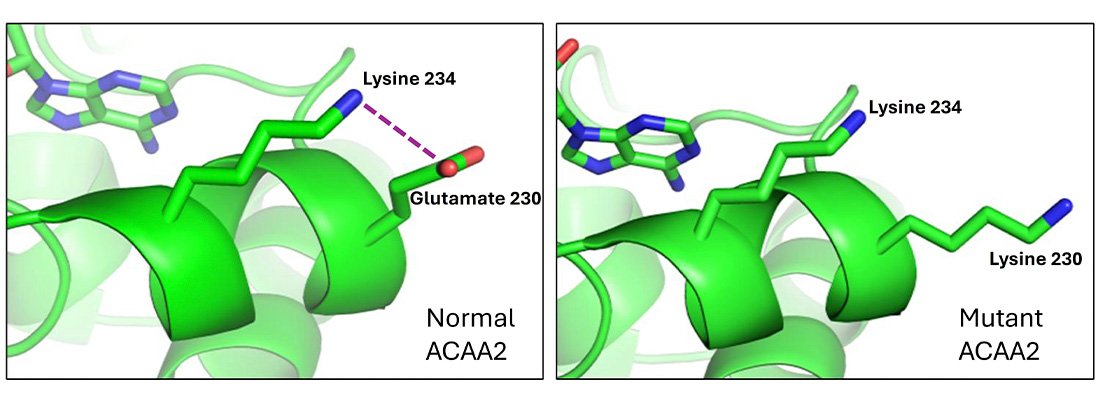
DALLAS – Dec. 09, 2025 – A rare genetic disorder discovered by UT Southwestern Medical Center researchers and their colleagues can cause brain damage from dangerously low blood sugar levels and liver damage in infants, along with variable body fat loss (lipodystrophy), fatty tumors, and metabolic complications in youth and adults. The study, published in The Journal of Clinical Investigation, found that the mutation on the ACAA2 gene can be inherited from a parent or occur spontaneously.
Abhimanyu Garg, M.D., Professor of Internal Medicine, Chief of the Section of Nutrition and Metabolic Diseases in the Division of Endocrinology, and the study’s lead investigator, suggested pediatricians treating patients with low blood sugar or hepatitis of unknown origins should consider testing for the genetic variant.

“We’ve found four families with the same genetic defect, and it is likely that clinicians will report more patients in the future,” Dr. Garg said. “Diagnosis of this disorder during infancy can not only prevent serious hypoglycemia but may also protect patients from hepatic (liver) dysfunction.”
The genetic variant causes a new subtype of familial partial lipodystrophy (FPL), a group of disorders that result in loss of body fat from the arms, legs, and hips and excess fat accumulation around the trunk, neck, and labia. Some patients develop fatty tumors called lipomas on their upper backs. Patients with FPL commonly develop metabolic dysregulation resulting in fatty liver disease, diabetes, and high blood fat levels. Dr. Garg has spent 30 years studying the genetic causes of FPL.
Dr. Garg explained that mitochondria, which act as powerhouses of human cells, use dietary fats consisting of chains of carbon molecules to produce energy for cellular functions. Several enzymes – including ACAA2, named after acetyl-coenzyme A acyltransferase 2 – are involved in breaking longer chains of fatty acids into shorter ones in a process called mitochondrial fatty acid oxidation.
In the newly discovered genetic variant, glutamic acid is replaced with another amino acid, lysine, within the ACAA2 enzyme that breaks down mostly medium-chain fatty acids.
Dr. Garg and his collaborators speculate the genetic disorder causes an overactivation of the ACAA2 enzyme. They also discovered a key diagnostic marker for the variant: elevated blood levels of long-chain acylcarnitines, which are derived from fatty acids.
Within the four families who were found to have the variant in the ACAA2 gene, six patients experienced transient hepatitis during infancy. Liver biopsies on two patients exhibited a buildup of fat deposits in liver cells, scarring, and damaged mitochondria.
“In one of the four families with the ACAA2 mutation, we saw low blood sugar levels during infancy,” Dr. Garg said. “One patient had such severe hypoglycemia that it resulted in brain damage.”
The fatty acid oxidation disorders prevent patients from generating ketone bodies, which can be used for energy when glucose is not available to fuel the brain.
With the discovery of the novel genetic variant, Dr. Garg sees the potential to develop disease models and test potential therapeutic interventions. He also questions whether other ACAA2 variants may be related to the development of diabetes, high blood triglycerides, or fatty liver in some patients.
“In the future, we expect dietary changes or novel therapies may emerge to lower long-chain acylcarnitine levels in affected individuals, which may reduce their morbidity,” Dr. Garg said.
Dr. Garg is an internationally recognized expert who provides clinical care and conducts research related to patients with rare lipid disorders and lipodystrophies. UTSW is one of 46 Rare Disease Centers of Excellence in 28 states and Washington, D.C., as recognized by the National Organization for Rare Disorders (NORD).
Other UTSW researchers who contributed to this study are Ralph J. DeBerardinis, M.D., Ph.D., Professor and Director of the Eugene McDermott Center for Human Growth and Development, Professor in Children’s Medical Center Research Institute at UT Southwestern (CRI) and of Pediatrics, and Director of the CRI Genetic and Metabolic Disease Program; Chao Xing, Ph.D., Professor in the McDermott Center, the Lyda Hill Department of Bioinformatics, and the Peter O’Donnell Jr. School of Public Health; Thomas Mathews, Ph.D., Assistant Professor in CRI and of Pediatrics; Purva Gopal, M.D., Associate Professor of Pathology; Diana Tomchick, Ph.D., Professor of Biophysics and Biochemistry; Anil Agarwal, Ph.D., Professor of Internal Medicine; and Xandria Johnson, B.S., Research Technician.
Dr. Garg holds the Distinguished Chair in Human Nutrition Research.
This study was funded by grants from the National Institutes of Health (NIH)/National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (R01-DK105448), NIH/National Cancer Institute (R35-CA220449), Southwestern Medical Foundation, the Intramural Research Program of the NIDDK, and the Cancer Prevention and Research Institute of Texas (RP240494).
About UT Southwestern Medical Center
UT Southwestern, one of the nation’s premier academic medical centers, integrates pioneering biomedical research with exceptional clinical care and education. The institution’s faculty members have received six Nobel Prizes and include 24 members of the National Academy of Sciences, 25 members of the National Academy of Medicine, and 13 Howard Hughes Medical Institute Investigators. The full-time faculty of more than 3,300 is responsible for groundbreaking medical advances and is committed to translating science-driven research quickly to new clinical treatments. UT Southwestern physicians in more than 80 specialties care for more than 143,000 hospitalized patients, attend to more than 470,000 emergency room cases, and oversee nearly 5.3 million outpatient visits a year.