
Structural Biology Lab
The Structural Biology Lab (SBL) provides access and assistance with the techniques of X-ray crystallography, small angle X-ray scattering and high-resolution imaging as well as single particle reconstruction in cryo-electron microscopy.
Getting Started
-
Review our pricing.
-
Study our resources.
-
Create an iLab account to schedule equipment.
Services
X-ray
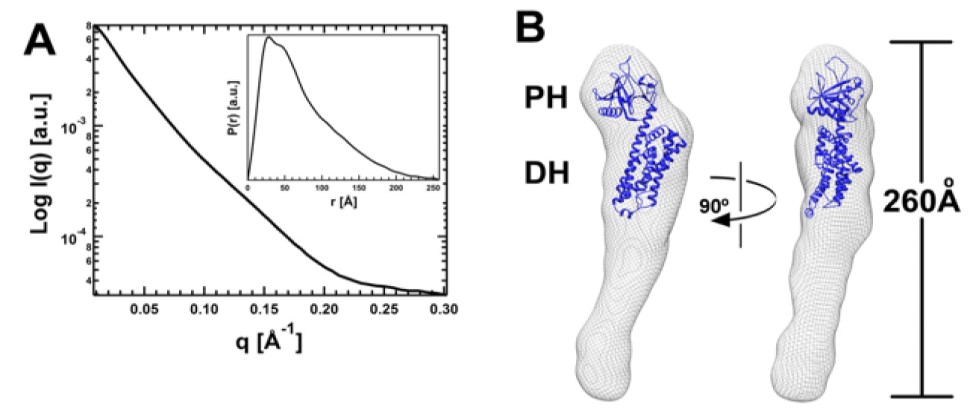
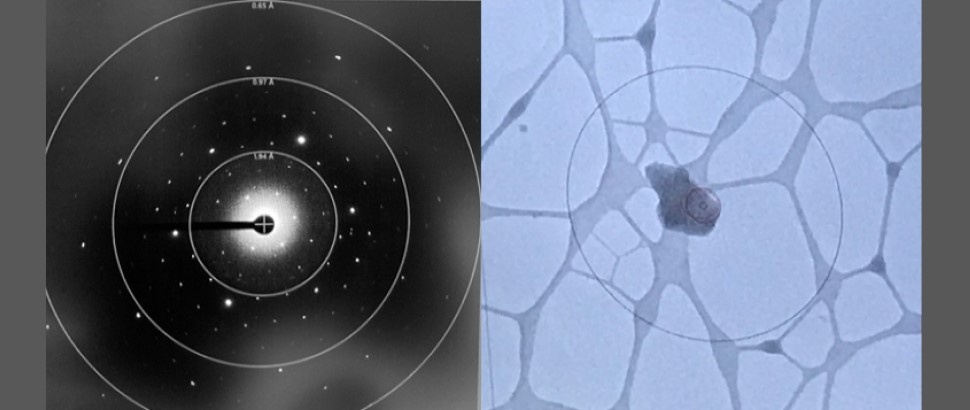
For X-ray, SBL offers assistance with crystal screening and optimization, in-house X-ray diffraction screening and data collection, synchrotron data collection at the Advanced Photon Source (APS) and other synchrotron sources for single crystals and small angle X-ray scattering (SAXS) measurements.
Single Particle Cryo-EM
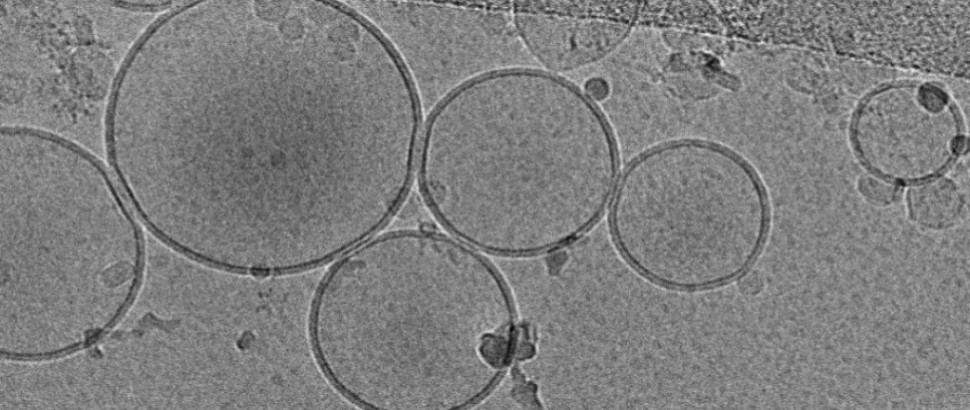
For single particle cryo-EM, SBL offers assistance with protein sample preparation, grid preparation for negative staining and cryo-EM, grid optimization, collection, processing, and analysis of data from in-house or remote microscopes, structure determination and validation.
MicroED
SBL is on-track to offer micro-crystal electron diffraction (MicroED) services for small molecule crystal screening and structure determination by the end of 2023.
About Our Funding
SBL is an Institutionally Supported Core and is supported through a combination of:
- institutional funds
- user fees
- federal and state grants, including one from the Cancer Prevention & Research Institute of Texas (RP220582).
Acknowledgement Policy
When publishing or presenting research when you were assisted by the staff or equipment in the SBL core lab, the acknowledgement policy is as follows:
- Cryo-EM: "For support with Cryo-EM studies, we thank the Structural Biology Lab at UT Southwestern Medical Center which is partially supported by grant RP220582 from the Cancer Prevention & Research Institute of Texas (CPRIT)."
- X-ray: "We thank the Structural Biology Lab at UT Southwestern Medical Center for support with X-ray crystallographic studies."
- If X-ray data was collected during the regularly scheduled APS synchrotron trips: "Results shown in this report are derived from work performed at Argonne National Laboratory (ANL), Structural Biology Center (SBC) at the Advanced Photon Source (APS), under Department of Energy Office of Biological and Environmental Research contract DE-AC02-06CH11357."
For presentation, simple acknowledgement of the SBL core lab (and specific staff member that assisted you) as well as simple acknowledgement of the SBC beamline at the APS is sufficient.
Acknowledgement of the CEMF for use of the microscope facility is also warranted if the Cryo-EM data was collected in their facility.
Location
North Campus Building ND
- Office Suite: ND10.214 and NB8.608
- X-ray Diffraction Facility: ND10.223
- Cryo-EM Sample Preparation Facility: ND8.226
Mailing Address
Structural Biology Lab (SBL)
Department of Biophysics
UT Southwestern Medical Center
5323 Harry Hines Blvd.
Dallas, TX 75390-8816
FedEx Address
Structural Biology Lab (SBL)
Department of Biophysics
UT Southwestern Medical Center
Room ND10.214
6001 Forest Park Rd.
Dallas, TX 75390

