Genetic mutation could worsen heart function in Duchenne muscular dystrophy patients
Those carrying a cystic fibrosis gene mutation could benefit from more aggressive and earlier cardiac interventions, study suggests
DALLAS – Nov. 4, 2020 – A mutation in the gene that causes cystic fibrosis may accelerate heart function decline in those with Duchenne muscular dystrophy (DMD), a new study by UT Southwestern researchers suggests. The findings, published online recently in the Journal of the American Heart Association, could help doctors develop new strategies to preserve heart function in this population, potentially extending patients’ lives.
DMD, caused by a mutation on the X chromosome, affects 1 of every 3,500 to 5,000 boys worldwide. This mutation results in the failure to produce dystrophin, a protein that protects muscle cells from damage, which in turn causes progressive muscle weakness. Although patients with DMD can suffer a variety of neuromuscular and lung complications, the cause of death – usually before age 35 – is typically cardiomyopathy, or weakness of the heart muscle.

All DMD patients eventually develop cardiomyopathy. But how early it develops and how progressive it manifests varies considerably, explains Pradeep Mammen, M.D., associate professor in the department of internal medicine’s division of cardiology at UTSW, who runs a cardiology clinic specifically for patients with DMD and other neuromuscular disorders.
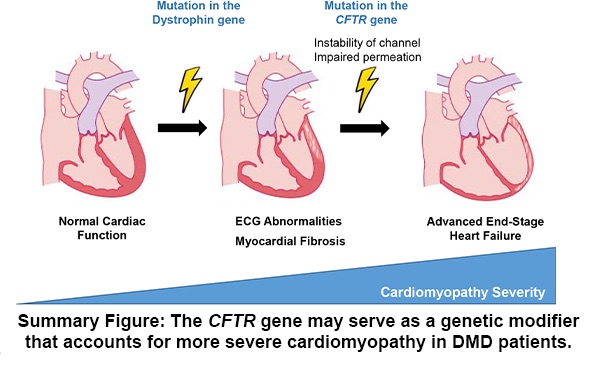
The reason for this variability has been unknown. However, Mammen and his colleagues suspected that it might result from an additional genetic variation, which may synergistically worsen heart function in DMD patients, accelerating the underlying cardiomyopathy.
To search for genetic variants that might have this effect, Mammen and his team recruited 22 male patients with DMD from their clinic and 12 female carriers, mostly mothers of patients. (Carriers were included in the study since they often also develop cardiomyopathy.) Cardiac function was assessed in 32 of these volunteers using either cardiac magnetic resonance imaging (cMRI), echocardiography, or cardiac computed tomography. Each volunteer also submitted blood to check for additional markers of cardiac function and to perform whole exome sequencing, a genetic test that reads all protein-making genes in the body.
In DMD patients with the worst cardiac function, the researchers identified a few candidate genes that might exacerbate their cardiomyopathy. But one in particular, a switch in a single unit of the cystic fibrosis transmembrane regular (CFTR) gene known as a “missense” mutation, stood out due to its role in heart cells. This gene, which causes cystic fibrosis when both the body’s copies carry a different characteristic mutation, is responsible for creating channels in heart cells that let bicarbonate in and regulate cell electrolyte levels.
DMD patients with the missense mutation in a single copy of this gene had a variety of markers of worse heart function compared with those who didn’t, including lower left ventricular ejection fraction, larger end-diastolic volume, and higher levels of a blood protein called N-terminal pro-B-type natriuretic peptide.
Although it is unknown how this CFTR mutation exerts its effects, Mammen says doctors who treat these patients might eventually test for this mutation to identify DMD patients who need more aggressive cardiac care at a younger age. Currently, DMD patients receive a range of interventions as cardiomyopathy develops and worsens, ranging from drugs (including ACE inhibitors, beta blockers, and mineralocorticoid receptor agonists) to left ventricular assist devices. Although patients typically begin receiving cardiac care in their teens to early 20s, patients carrying the CFTR mutation may benefit from starting aggressive care earlier to prevent heart damage.
“Even with new strategies to treat these patients on the horizon, such as genome editing that could convert DMD to a less severe form known as Becker’s muscular dystrophy, cardiomyopathy will continue to be a patient’s most serious and life-ending consequence,” says Mammen, who holds the Alfred W. Harris, M.D., Professorship in Cardiology. “Finding ways to help preserve heart function over time could offer new hope for patients with DMD.”
Other UTSW researchers who participated in this study include Xuan Jiang, Yanqiu Shao, Faris Araj, Alpesh Amin, Benjamin Greenberg, Mark Drazner, and Chao Xing. Drazner holds the James M. Wooten Chair in Cardiology and Greenberg is a Cain Denius Scholar in Mobility Disorders.
This study was supported by a National Institutes of Health Research Project Grant (NIH R01HL102478) and the National Institutes of Health funded UT Southwestern Sen. Paul D. Wellstone Muscular Dystrophy Cooperative Research Center (NIH U54HD087351).
About UT Southwestern Medical Center
UT Southwestern, one of the nation’s premier academic medical centers, integrates pioneering biomedical research with exceptional clinical care and education. The institution’s faculty members have received six Nobel Prizes and include 24 members of the National Academy of Sciences, 25 members of the National Academy of Medicine, and 13 Howard Hughes Medical Institute Investigators. The full-time faculty of more than 3,300 is responsible for groundbreaking medical advances and is committed to translating science-driven research quickly to new clinical treatments. UT Southwestern physicians in more than 80 specialties care for more than 143,000 hospitalized patients, attend to more than 470,000 emergency room cases, and oversee nearly 5.3 million outpatient visits a year.